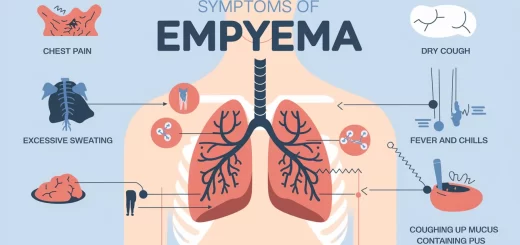
Interstitial Lung Diseases stages, types, treatment and How serious is interstitial lung disease
Interstitial Lung Diseases (ILDs) are a group of lung disorders that cause inflammation and scarring (fibrosis) of the interstitium—the tissue surrounding the air sacs (alveoli) in the lungs. This scarring makes it harder for oxygen to pass into the bloodstream, leading to breathing difficulties.
Interstitial Lung Diseases
Interstitial lung diseases are a group of diseases that lack parallelism between signs and symptoms. Diagnosis requires a high suspicion index in many cases. ILD affects the lung interstitial, which is the tissue and space around the air sacs of the lungs; it concerns alveolar epithelium, pulmonary capillary endothelium, and basement membrane perivascular and peri lymphatic tissues.
Nomenclature
- Diffuse parenchymal lung disease (DPLD) (common).
- Alveolar filling.
- The term interstitial is misleading since most of these disorders are also associated with extensive alterations of alveolar and airway architecture.
Prevalence
More than 200 diseases can result in ILD. 31.5 per 100.000 men, 26.1 per 100.000 women.

Interstitial Lung Diseases
Pathophysiology
Acute injury to the pulmonary parenchyma leads to chronic interstitial inflammation, then to fibroblast activation and proliferation and finally progressing to the end point of pulmonary fibrosis and tissue destruction.
In the early stages due to the deposition of fibrous tissue in the interstitium Hypoxia but the diffusion of CO2 is higher than O2, so hypocapnia or normocapnia will occur (Type 1 respiratory failure).
Type 2 respiratory failure occurs in COPD patients or any obstructive lung disease with hypercapnia. In late stages, these patients will be hypercapnic due to extensive fibrosis in the interstitium.
Spectrum of DPLD
It may be classified as:
1. Idiopathic:
- Idiopathic Pulmonary fibrosis
- Non-specific Interstitial pneumonia.
- Desquamative interstitial pneumonia.
- Respiratory bronchiolitis interstitial pneumonia.
- Acute interstitial pneumonia.
Cryptogenic interstitial pneumonia: is the migrating pneumonia,
- It appears in CT as patches at the periphery migrating from one site to another so the best tool to diagnose it is percutaneous needle biopsy CT guided.
- If another patient comes with the same lesion but every CT appears at the same site not migrating and from the history it will be clear that there is obstruction close to the airway and cause wheezes (Peribranchial), The investigation will be bronchoscopy and depending on the history.
• Lymphocytic interstitial pneumonia.
2. Secondary to a specific cause:
- Connective tissue diseases such as SLE, RA, and scleroderma.
- Radiotherapy.
- Infections e.g, tuberculosis (may develop from the residue of active infection of any type)
- Malignancy.
- Drug-related e.g., bleomycin, methotrexate, amiodarone, nitrofurantoin.
- Occupational exposures e.g.. farmer’s lung,
- byssinosis, hypersensitivity pneumonitis.
3. Granulomatous DPLD
- Sarcoidosis.
- Acute interstitial pneumonia.
- Cryptogenic interstitial pneumonia.
- Lymphocytic interstitial pneumonia.
- Drug-related e.g., bleomycin, methotrexate, amiodarone, nitrofurantoin.
- Occupational exposures e.g., farmer’s lung, byssinosis, hypersensitivity pneumonitis.
- Pulmonary Langerhans cell histiocytosis (PLCH), previously known as Histiocytosis X.
4. Others
- Eosinophilic pneumonia.
- Lymphangioleiomyomatosis (LAM).
The diagnosis of IIPs requires the exclusion of known causes of interstitial lung disease (such as drug or inhalational exposure and Collagen vascular diseases).
Coexisting Patterns: Most patients with a chronic IIP can be given a single radiologic-pathologic diagnosis. However, multiple clinical-pathologic and/or HRCT patterns may be found in the same patient. When coexisting patterns occur, Multi-Disciplinary Discussion (MDD) may determine the clinical significance of individual patterns.
Diagnostic approach:
1. History
- Onset: Chronic: e.g. IIPs, sarcoidosis. Acute or subacute eg. EAA, drug-induced.
- Sex: eg lymphangioleiomyomatosis
- Age: younger (sarcoidosis), IPF (late middle age).
- Occupational history: listing of lifelong employment.
- Environmental exposure: pets (especially birds), air conditioners, humidifiers & evaporating cooling systems.
- Smoking history: alter ILD.
- Family History: e.g. IPF, sarcoidosis.
- Systematic diseases.
- ➤ Current/Previous medications.
2 Clinical picture
- Asymptomatic with abnormal findings on radiographs or pulmonary functions.
- Typically present with progressive exertional dyspnoea and/or persistent nonproductive cough.
- Symptoms are nonspecific, subtle, and slowly progressive.
- Environmental exposures (inhaled allergens, dust), concomitant systemic illnesses such as collagen vascular disease Who are at risk?
- Clubbing (most common in IPF).
- Dry bibasilar crepitations / Squeaks.
- Signs of hypoxemia and respiratory failure (cyanosis) which occur late in the course of the disease.
- Signs of right heart failure (LL edema, hepatomegaly, congested neck veins) occur late in advanced disease.
- Other unusual chest symptoms:
- Hemoptysis e.g., alveolar hemorrhage syndromes, pulmonary vascular diseases & chronic mitral valve diseases.
- Pleuritic chest pain→ e.g., collagen vascular illness.
- Wheezing e.g., Churg-Strauss syndrome & EAA.
3. Investigations
1. Initial laboratory:
- Complete blood count with differential, ESR.
- Renal & liver function studies.
- Antinuclear antibodies, antineutrophil cytoplasmic antibody, rheumatoid factor.
- Urine analysis.
N.B: Routine tests→ nonspecific.
2. Pulmonary functions: Usually a restrictive abnormality
- Normal FEV1/FVC ratio.
- Reduced FEVI, VC, and TLC.
- Impaired diffusing capacity, (decreased DLCO).
- Mixed restrictive and obstructive PFT abnormality if DPLD is superimposed on COPD or at the end stage.
- Arterial hypoxemia at rest/exercise.
3. Radiology:
- Chest x-ray: normal 10%.
- High-resolution CT chest (HRCT): is the gold standard imaging modality for patients with ILD showing.
- Reticular or reticulonodular pattern.
- Alveolar shadow.
- Ground glass opacities.
- Honeycombing and traction bronchiectasis.
- Interlobar septal thickening.
4. Arterial blood gases:
- Early: hypoxia, hypocapnia
- Late: hypoxia, hypercapnia.
5. Bronchoalveolar-lavage: (Narrow the differential diagnosis)
- Diagnostic infectious causes, occupational exposures, malignancy & pulmonary alveolar proteínosis.
- Differential cell count of BAL.
- Sarcoidosis: lymphocytosis, T-helper cells with a high CD4/CD8 ratio.
- Hypersensitivity pneumonitis: marked T lymphocytosis (CD8) with low CD4/CD8 ratio
- IPF: increases in neutrophils and eosinophil.
6. Lung biopsy:
Transbronchial lung biopsy:
- Minimally invasive, performed at the same time as BAL. Useful in the diagnosis of some ILDs eg. sarcoidosis, infection or lymphangitis carcinomatosis.
- The use of Endobronchial ultrasound (EBUS) is useful in reaching the target area and decreasing the risk of bleeding and pneumothorax.
Percutaneous needle biopsy:
- Under local anesthesia with fluoroscopic, ultrasound or CT guidance,
- Precise sampling from localized areas of abnormality
- A small amount of tissue.
- Risk of bleeding & pneumothorax.
Surgical lung biopsy:
- Technique: Video-assisted thoracoscopic surgery (VATS) or limited thoracotomy.
- Diagnostic yield (92%), morbidity (2.5%) & mortality (0.3%).
- Histopathologic evaluation has become the basis for the classification of the ILD, prognostic information, and guide therapy,
- Severe end-stage DPLD or significant concomitant illness may be wise not to proceed to SLB.
The Idiopathic Pulmonary Fibrosis (IPF)
- IPF is defined as a specific form of chronic, progressive fibrosing interstitial pneumonia of unknown cause, occurring primarily in older adults, limited to the lungs, and associated with the histopathologic and/or radiologic pattern of usual interstitial pneumonia (UIP).
- Prevalence is estimated to be slightly greater in men than in women, prevalence peaks at 65-79 years of age.
Suggested risk factors for IPF
Although the cause of IPF is unknown, several risk factors have been suggested:
- Cigarette smoking: Strongly associated with IPF.
- Genetic factors: Familial pulmonary fibrosis accounts for 5% of the total population with IPF.
- Environmental pollutants: Associated with an increased risk of IPF. Exposure to metal and wood dust, farming, raising birds, hairdressing, stone cutting/polishing, and exposure to livestock, vegetables or animal dust.
- Infection: Epstein-Barr virus “in 96%” of hepatitis C.
- Gastroesophageal reflux (GER) disease: Proposed cause of repeated microinjury.
- Diabetes mellitus?!
Clinical presentation of IPF:
symptoms and clinical signs of IPF often appear gradually and include:
- Slowly progressing dyspnea.
- Non-productive cough.
- Dry, inspiratory bibasilar “Velcro-like” fine crepitations.
- Clubbing of fingers
- Abnormal pulmonary function test results, with evidence of restriction and impaired gas exchange. IPF is a debilitating restrictive lung disease, affecting the patient’s daily routine physical activities. IPF disease progression is inevitable yet unpredictable. Acute exacerbations of IPF often have no identifiable cause and result in high mortality.
Treatment
To date, there is no therapy proven to improve survival or otherwise significantly modify the clinical course of IPF. As such, it is recommended that all patients be considered for recruitment to high-quality clinical trials of therapy and/or for lung transplantation if appropriate.
- Referral to a transplant center for lung transplantation should be made if the disease is advanced (DLCO <40% predicted) or progressive (>10% decline in FVC or ≥ 15% decline in diffusion during 6 months of follow-up).
- Best supportive care should be considered a specific and important treatment strategy in all patients with IPF. It is a proactive approach to symptomatic treatment and may include oxygen therapy and pulmonary rehabilitation.
- Pneumococcal and influenza vaccination should not be overlooked.
- Treatment Varies according to disease.
- Drugs: corticosteroids, other immunosuppressive agents e.g., azathioprine, cyclophosphamide, anti-fibrotic agents e.g., colchicine.
- New anti-fibrotic agents: pirfenidone, nintedanib (tyrosine kinase inhibitor).
- Antioxidants e.g., glutathione, N-acetyl cysteine.
- Supplemental oxygen.
- Pulmonary rehabilitation
- For end-stage disease, lung transplantation remains a restricted, but often effective option
ILDs are a group of complex disorders usually presenting with progressive dyspnea, restrictive pulmonary physiology, and an abnormal chest radiograph. Because of overlapping signs and symptoms, the evaluation of these patients is often frustrating; the key to understanding and correctly diagnosing ILD is the development and utilization of a disciplined evaluation process. The use of such standardized, logical evaluation will yield a diagnosis in the majority of patients with ILD.
You can subscribe to Science Online on YouTube from this link: Science Online
You can download Science Online application on Google Play from this link: Science Online Apps on Google Play
Complications of Bronchogenic carcinoma, Prevention and risk factors of lung cancer
Bronchogenic Carcinoma types, stages, symptoms and diagnosis
Pneumothorax causes, types, treatment, Who is at risk for pneumothorax?
Pleural disease symptoms and treatment, What is the most common pleural disease?
Bronchiectasis causes, Is a bronchiectasis serious?, What is the best medication for bronchiectasis?
Suppurative Lung Diseases, Stages of Lung Abscess, Does a lung abscess require surgery?
Tuberculosis symptoms, What causes TB disease? and Can TB be transmitted through saliva?
Inflammation of the lung parenchyma, What causes lung parenchyma?, Is lung parenchyma dangerous